Pathogene Alzheimer-Kaskade wird aktiviert durch fehlerhafte Noradrenalin-Signalisierung

In präklinischen Experimenten an der University of Alabama at Birmingham-Forscher haben gezeigt, eine Taste fehlt Stück von der Alzheimer-Krankheit puzzle. Erlaubt, dass die proof-of-concept—Experimente unter Verwendung einer vorhandenen Droge, die drastisch reduziert die Alzheimer-Pathologie und Symptome in beiden Maus-Modellen, potenziell bietet eine sofortige Behandlung für diese verheerende Krankheit.
Die Forschung wurde heute veröffentlicht in der Fachzeitschrift Science Translational Medicine. Sie enthalten menschliche Gehirn Gewebe-Analyse-und längs-klinische Daten, unterstützt die in-vivo-Maus-Modell-Daten.
„Unsere Studie liefert Einblicke in die translationale Mechanismen der amyloid-beta-protein-Toxizität, die möglicherweise starke Implikationen für die zukünftige Medikamentenentwicklung“, sagte Qin Wang, M. D., Ph. D. „Es gibt eine amyloid-beta – /G-protein-gekoppelt-rezeptor-Interaktion, stellt ein attraktives, Krankheit-spezifisches therapeutisches zielmolekül für die Alzheimer-Krankheit.“
Interessanterweise ist der pathologische Mechanismus gefunden, könnten auch eine Erklärung für das scheitern der zahlreichen Alzheimer klinische Studien, dass eine gezielte Reduktion der Täter bei der Alzheimer-Krankheit—amyloid-protein-Ablagerungen im Gehirn.
An der UAB School of Medicine, Wang ist professor in der Abteilung für Zell -, Entwicklungs-und Integrative Biologie.
Es ist weithin anerkannt, Wang sagt, dass Ablagerungen von amyloid-beta-oligomere im Gehirn fungiert als trigger, um zu veranlassen, pathologische Veränderungen des tau-proteins, und das veränderte tau-protein ist die Kugel, die Ziele und tötet Nervenzellen bei der Alzheimer-Krankheit. Jedoch, der Weg der Verbindung dieser beiden war unbekannt.
Wang und Kollegen haben festgestellt, dass amyloid-beta-oligomere hijack Noradrenalin-Signaltransduktion an Nervenzellen im Gehirn, die fälschlicherweise leitet dieses signal zur Aktivierung einer kinase namens GSK3-beta. Dass aktivierte-kinase-Enzym, in turn, hyper-phosphoryliert tau-protein, so dass es toxisch für die Neuronen.
Diese Neuverkabelung des Noradrenalin-Signalisierung stattfindet, in einem Zell-Membran-rezeptor auf der Oberfläche von Neuronen, die so genannten alpha-2A-adrenergen rezeptor. Dieser rezeptor ist Teil einer großen Familie der G-protein-gekoppelte Rezeptoren, die erkennen, Moleküle außerhalb der Zelle, und aktivieren Sie dann ein internes signal, das bewirkt, dass eine zelluläre Reaktion. Während einer bestimmten Konzentration von amyloid-beta-oligomere aktivieren kann, GSK3-beta, die Anwesenheit von Noradrenalin stark sensibilisiert, dass die Aktivierung von bis zu zwei Größenordnungen, Wang und Kollegen gefunden.
So, die UAB-Forscher spekulieren, dass nanomolaren Konzentrationen von amyloid-beta oligomers in human brains induzieren eine pathogene GSK3-beta/tau-Kaskade in den frühesten Stadien der Alzheimer-Krankheit. Diese Theorie besagt, warum mehrere klinische Studien zum Abbau von amyloid-beta-oligomeren in der Alzheimer-Krankheit Patienten, die versagt haben, Sie nicht zu einer Verringerung der amyloid-Ebenen zu solch niedrigen Konzentrationen.
Einzelheiten der Studie
Der alpha-2A-adrenergen rezeptor normalerweise funktioniert das so—es hat eine Bindungsstelle für den neurotransmitter Noradrenalin, und diese Bindung aktiviert ein Signal-Prozess, mobilisiert das Gehirn und Körper auf Aktion. Die UAB-Forscher fanden heraus, dass die amyloid-beta-oligomere binden an eine separate Website auf der alpha-2A-adrenergen rezeptor, die sich von der Website für Noradrenalin binden. Dieser leitet die pathologisch-hijacking.
Eine solche Bindung an eine zweite Seite aufgerufen wird allosterische Bindung. In der G-protein-gekoppelten Rezeptoren durch allosterische Liganden sind bekannt, oft verändern Signaltransduktion von der rezeptor als Teil der normalen Physiologie. Nachdem die Forscher erkannten die allosterische Bindung, die Sie gesucht, um zu sehen, welche kinase-Aktivierung durch die Bindung, die ist, wie Sie identifiziert GSK3-beta.
Einige klinische Daten unterstützt diesen Mechanismus. Die Forscher fanden heraus, dass alpha-2A-adrenergen rezeptor aus der post-mortem-präfrontalen cortexbereiche von Alzheimer-Patienten hatte einen signifikanten Anstieg der alpha-2A-adrenergen rezeptor-Aktivität, im Vergleich mit nicht-dementen, low-Pathologie kontrolliert. Auch epidemiologische Analyse von Fällen aus der National Alzheimer ‚ s Coordinating Center zeigte, dass die Einnahme des Arzneimittels Clonidin—eine Aktivierung von alpha-2A-adrenergen rezeptor verwendet, um den Blutdruck zu senken—verschlechtert die kognitive Funktion bei Patienten mit kognitiven Defiziten. Darüber hinaus die Nebenwirkungen von Clonidin stärker waren bei Patienten mit schwerer Demenz. Clonidin Nutzung hatte keinen Einfluss auf Patienten mit normaler Kognition.
Wang und Kollegen untersuchten eine bestehende Drogen—idazoxan—in einem Maus-Modell der Alzheimer-Krankheit. Idazoxan ist ein alpha-2A-adrenergen rezeptor-antagonist, wurde unter Untersuchung in klinischen Studien für Depressionen. Die Hypothese war, dass idazoxan Blockade von alpha-2A-adrenergen rezeptor in Anwesenheit von amyloid-beta-Pathologie zeigen würde therapeutischen Möglichkeiten. Das war getragen, die in Alzheimer-Modell-Mäusen.
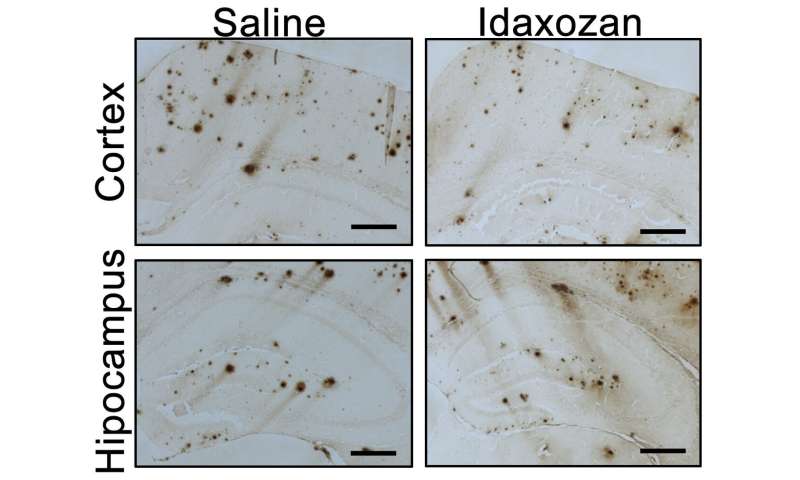
Die Mäuse wurden behandelt mit idazoxan für acht Wochen beginnend im Alter von 8 Monaten, ein Punkt, wenn amyloid-beta-plaques sind bereits in die Gehirn-und alpha-2A-adrenergen rezeptor zeigt verstärkte Aktivität. Im Vergleich mit den Kontrollen, die UAB-Forscher fanden heraus, dass: 1) Idazoxan Umgekehrt hyperactivation von GSK3-beta in der Maus-Gehirne, geben zusätzliche Unterstützung für die wichtige Rolle von alpha-2A-adrenergen Rezeptors in der Vermittlung des amyloid-beta-induzierte Aktivierung von GSK3-beta in vivo; 2) In der Großhirnrinde von idazoxan-behandelt die Alzheimer-Modell-Mäusen das Ausmaß der amyloid-beta-Belastung geringer war, zeigen, dass die Blockade der alpha-2A-adrenergen rezeptor, verlangsamt das Fortschreiten der amyloid-beta-Pathologie; 3) Idazoxan Behandlung verringerte sich die Dichte der entzündlichen microglial Zellen, was auf eine Verringerung der Nervenentzündung; 4) Idazoxan Behandlung reduziert tau hyper-Phosphorylierung, was darauf hindeutet, dass die blockade des alpha-2A-adrenergen Rezeptors gelindert amyloid-beta-induzierte tau-Pathologie; und 5) Idazoxan-behandelt die Alzheimer-Modell-Mäusen durchgeführt fast so gut wie normale Mäuse, und deutlich besser als unbehandelte Alzheimer-Modell-Mäusen in beiden tests für die kognitive Funktion.
„Diese Daten zusammenfassend zeigen, dass eine Blockade von Noradrenalin-Signal durch den alpha-2A-adrenergen rezeptor ist eine wirksame Strategie zur Verbesserung der pathologischen und kognitive Defizite im Zusammenhang mit amyloid-beta,“ Wang sagte.
„Der alpha-2A-adrenergen rezeptor-Blocker wie idazoxan wurde entwickelt für den Einsatz bei anderen Erkrankungen eingesetzt werden, und die Wiederverwendung von diesen Medikamenten könnte ein potenziell wirksames, leicht verfügbar Strategie für die Alzheimer-Behandlung“, sagte Wang. „Zudem weisen unsere Daten darauf hin, dass die amyloid-beta – /alpha-2A-adrenergen rezeptor-Interaktion ist eine attraktive, Krankheit-spezifisches therapeutisches Ziel für Alzheimer, weil der alpha-2A-adrenergen rezeptor – /GSK3-beta/tau-Kaskade kann aktiviert werden, nur in Anwesenheit von amyloid-beta-oligomeren.“