Die coronavirus-Genom ist wie ein Versandetikett, mit der Epidemiologen verfolgen, wo es war

Nach der Corona-Virus ist verbreitet in der Bevölkerung—und die Vorwegnahme Ihrer nächsten bewegen—ist ein wichtiger Teil der öffentlichen Gesundheit Reaktion auf die neue Krankheit, die vor allem seit containment ist unsere einzige Verteidigung so weit.
Schauen Sie einfach nur auf eine infizierte person nicht sagen, wo Ihre version des coronavirus kam, und SARS-CoV-2 nicht über ein bar-code, den Sie Scannen können, um Ihnen zu erlauben, zu verfolgen, seine Reise-Geschichte. Jedoch, seine genetische Sequenz ist fast so gut, einen Einblick in, wo Sie den virus hat.
Das Genom eines Organismus ist die vollständige genetische Anweisungen. Sie können denken, ein Genom als ein Buch, die Wörter aus Buchstaben. Die einzelnen „Buchstaben“ des Genoms ist ein Molekül namens ein Nukleotid—in Kurzschrift, A, G, C, T oder U.
Mutationen können immer dann auftreten, wenn das virus repliziert sein Genom, so dass im Laufe der Zeit Mutationen in das virale Genom. Zum Beispiel, anstelle von „Wort“ KATZE, das neue virus hat GAT. Das virus führt diese geringfügige änderungen, da es sich von einer person zur nächsten host.
Diese Mutationen Verhalten sich wie ein Pass-Stempel. Egal wo Sie gehen weiter, zurück-Stempel in Ihren Pass zeigen noch, wo Sie gewesen sind.
Molekular-Genetiker wie wir diese Informationen verwenden können, um zu konstruieren Stammbäume für die coronavirus. Das ermöglicht es uns, verfolgen Sie die Routen, die das virus hat, gereist durch Raum und Zeit und starten Sie zu beantworten Fragen wie, wie schnell und einfach das tut es verbreitete sich von einer person zur anderen?
Individuelle Patienten-Daten helfen, malen ein großes Bild
Online-Datenbanken gesammelt haben SARS-CoV-2 die genomische nukleotidsequenzen seit Mitte Dezember. Wenn ein patient positiv getestet für die SARS-CoV-2, a lab können bestimmen, dass die Genom-Sequenz des infizierenden virus und hochladen. Ab Ende April, mehr als 1.500 Genom-Sequenz-Proben wurden hinterlegt in GenBank, eine öffentlich zugängliche Datenbank wird von der National Institutes of Health, und mehr als 3.000 in GISAID, die open-access Global Initiative on Sharing All Influenza Data.
Da jede Sequenz aus einem Patienten, der an einem bestimmten Ort in der Welt, diese virale Genom-Sequenzen, die es Wissenschaftlern ermöglichen, Sie zu vergleichen und zu verfolgen, wo der virus wurde. Desto ähnlicher sind sich die Sequenzen von zwei bestimmten Viren sind, die eng verwandt Sie sind, und die mehr vor kurzem Sie haben einen gemeinsamen Vorfahren. Die ersten SARS-CoV-2 die genomische Sequenz hochgeladen, um die GISAID Webseite wurde gesammelt von einem Patienten im frühen Dezember 2019.
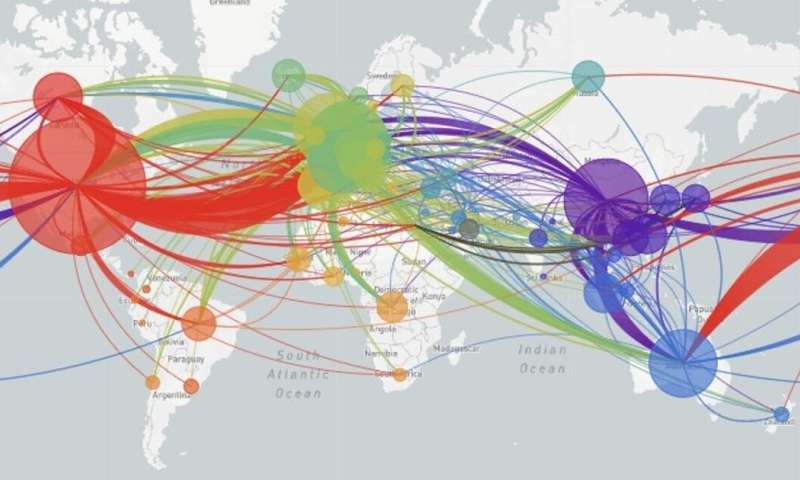
Natürlich, die virale Mutationen selbst nicht sagen Forscher, in welchem Land Sie passiert in. Aber da die Datenbanken aufnehmen, in denen bestimmte Muster von Mutationen beobachtet wurden, können die Wissenschaftler bestimmen den Weg, dass jeder viralen Belastung genommen hat. Auf der Weltkarte verfolgt die Bewegung des virus auf der ganzen Welt.
Die erfassten Daten von tausenden von Patienten zeigen, dass SARS-Cov-2 entstand in Wuhan, China und verbreitete sich von dort auf den rest der Welt.
Gebäude, Karten von Sequenzen
Die genetischen Daten können spielen eine große Rolle beim knacken der öffentlichen Gesundheit Geheimnisse, wie die Corona-Virus verbreitet hat, durch die Vereinigten Staaten.
Zum Beispiel, ein Reisender aus Wuhan Ankunft in Seattle am Jan. 15 und positiv getestet für das virus auf Jan. 20.
Auf Feb. 28, Wissenschaftler sequenzierten eine virus-Probe aus einem amerikanischen Patienten in Seattle und fand seine mutation Signatur aufeinander abgestimmt, dass sich der virus von der Wuhan Reisenden, plus drei neue Mutationen. GISAID hat errechnet, dass die Mutationsrate bei etwa 0.45 Mutationen pro Genom pro Woche—also drei Mutationen zwischen Jan. 20 Fall und die Feb. 28 Fall passt dieser Satz.
Basierend auf den drei neuen Mutationen, diese version des virus wurde die Multiplikation unentdeckt für etwa fünf Wochen in der Gegend von Seattle. Da jeder infizierten person anstecken können mehrere andere Menschen ohne irgendwelche Symptome selbst, das virus könnte haben sich auf die mehr als 100 Menschen in fünf Wochen.
Mit Hilfe der Genom-Sequenzen zu verknüpfen, das virus aus dem Jan. 15 Reisende aus Wuhan mit der in Washington ansässige patient, der von Ende Februar gewarnt, Washington state Beamten, dass der virus war still, die Runde durch die Bevölkerung. Dieses unerkannte Verbreitung des virus in Seattle und anderswo ist einer der primären Gründe der öffentlichen Gesundheit Beamten fordern die öffentlichkeit, um zu Hause zu bleiben, so viel wie möglich.
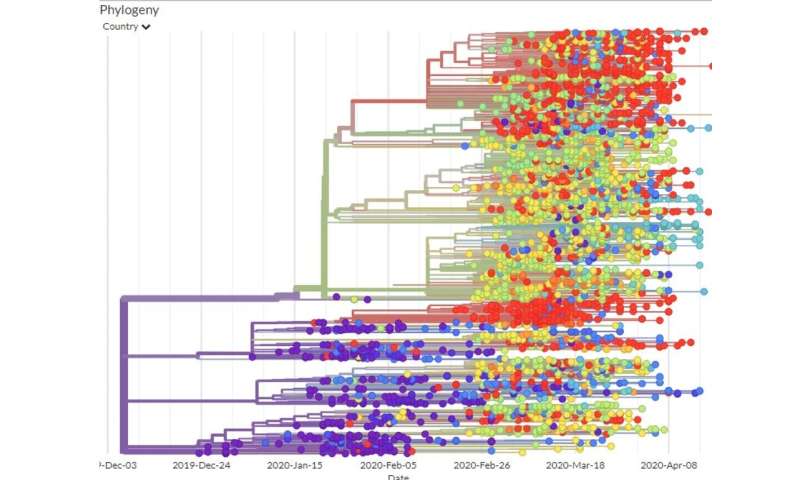
Eine andere Studie detailliert den Weg, den das virus nahm, als er zog von Wuhan nach Shanghai nach Deutschland, nach Italien, Mexiko, verstauen in infizierten Reisenden. Diese Studie verfolgt infizierten Personen und verglichen Ihre viralen genomischen Sequenzen. Da könnten Forscher vergleichen die virale Mutationen zu denen in bekannten Orten zu bestimmten Zeiten, Sie waren in der Lage, eine Karte der phylogenetische Baum—Stammbaum, der zeigt, wie die verschiedenen virus-Genom-Sequenzen verwandt sind.
Unter Verwendung der GISAID geschätzte Mutationsrate und der phylogenetischen Baum, denken Wissenschaftler das erste mal die Corona-Virus infiziert, eine person wahrscheinlich aufgetreten in Wuhan im November oder Anfang Dezember 2019.
Wenn der virus hatte es schon viel länger, die Viren der ersten bekannten Patienten hätten eine größere Vielfalt von Mutationen, als Sie es Taten.
Immer noch verfolgen und das lernen von den Sequenzen
Die Analyse der viralen genomischen Sequenzen, wird auch weiterhin ein wertvolles Werkzeug für die Verfolgung und Eindämmung der Ausbreitung von SARS-CoV-2.
Zum Beispiel die Sequenzierung des Genoms eines virus von einem neu infizierten Patienten könnte Sie sagen, wenn es ist ein virus, der kursiert in der Gegend für eine Weile, oder wenn es eine neue Einführung von anderswo.
Jemand, der gewesen war, in Nord-Italien vor Reisebeschränkungen wurden, brachte das virus nach Island. Das erste Ausbruch eingedämmt wurde ziemlich schnell, aber dann neue Formen des virus eingeführt wurden von anderswo in Europa.
Eine neue Studie pending peer-review zeigt, dass Kalifornien auch hatten mehrere Einführungsveranstaltungen mit verschiedenen viralen Linien. Für Kalifornien, ist die Kenntnis der Häufigkeit von Neueinführungen wäre ein wichtiger Faktor, um als Beamte entwickeln Möglichkeiten, um das virus enthalten.