Akute chikungunya-Infektion untersucht auf molekularer Ebene, die in der brasilianischen Patienten

Computational tools angewandt, um die Biologie revolutionieren die Studie von dem, was passiert in den Zellen während einer Infektion, hilft Wissenschaftlern zu verstehen, krankheitsmechanismen und einen Beitrag zur Identifikation potentieller therapeutischer targets.
Ein Beispiel ist eine Studie, veröffentlicht in PLOS-Krankheitserregern , die beschreibt, wie Brasilianische Forscher analysierten Zellen im Blut von infizierten Patienten mit dem chikungunya-virus. Mit Hilfe von Techniken wie Analyse komplexer Netzwerke, künstliche Intelligenz und maschinelles lernen, in der Gruppe identifizierten gensignaturen im Zusammenhang mit der Krankheit—sets von Genen, deren expression verändert sich durch die Interaktion mit dem virus. Sie untersucht die Rolle, die in den Zellen durch die beteiligten Gene und bestimmt die Bedeutung dieser Gene für die Bemühungen um die Bekämpfung des virus.
Durchgeführt in Brasilien, die Forschung wurde unter Leitung von principal investigator Helder Nakaya, professor an der University of São Paulo School of Pharmaceutical Sciences (FCF-USP). Forscher an der gleichen Universität Biomedical Science Institute (ICB-USP) und seine Ribeirão Preto Medical School (FMRP-USP) sowie den Kollegen am Butantan Institut und der Öffentlichen Gesundheit-Central Laboratory of Sergipe, unter anderem, auch dazu beigetragen.
„Wir haben auch festgestellt, eine Reihe von Genen, die zeigen, dass während der akuten phase, ob der patient wahrscheinlich entwickeln chronische Gelenkschmerzen [Gelenkschmerzen und-Entzündungen], eine relativ häufige Erkrankung in infizierten Menschen mit chikungunya. Jedoch, dieses Ergebnis muss noch bestätigt werden, durch die zukünftige Forschung basierend auf einer größeren Anzahl von Proben,“ sagte Nakaya.
Der Artikel beschreibt die Ergebnisse der Analysen mit Hilfe der Blutproben von 39 Probanden, geboren in Sergipe, einem Staat im Nordosten Brasiliens, und infiziert während der 2016-Epidemie. Diese Ergebnisse wurden verglichen mit Daten einer Kontrollgruppe bestehend aus 20 nicht-infizierten Probanden aus der gleichen region.
Der erste Schritt war eine Analyse der Proben “ transkriptoms umfasst alle Moleküle der messenger-RNA (codiert Proteine) als auch nicht-kodierende RNAs (die keine Proteine produzieren, sondern führen eine regulatorische Rolle in der Genom), ausgedrückt in den roten Blutkörperchen, weißen Blutkörperchen und Blutplättchen. Durch die Quantifizierung der Transkript Level in den Proben, konnten die Forscher Messen die Aktivität von 20.000 Genen und bestimmen, ob Ihre expression erhöht oder verringert werden, während der Infektion; diese Ergebnisse wurden auch im Vergleich mit den Ergebnissen für die Kontrollgruppe.
„Wir konzentrierten uns auf die protein-kodierenden Genen [die express-boten-RNA], da Ihre Rolle ist leichter zu interpretieren. Es ist relativ einfach, zu wissen, ob Sie Kodieren-Zell-Rezeptoren oder Transkriptionsfaktoren, zum Beispiel. Auf diese Weise waren wir in der Lage, um zu verbessern unser Verständnis der Pathogenese von chikungunya—, wie das virus befällt Zellen und die Verteidigungssysteme sind aktiviert in Reaktion auf eine Infektion,“ sagte Nakaya.
Ihre Analyse zeigte den Mechanismus, mit dem Zellen des Immunsystems, trigger-entzündliche Prozesse, das virus zu eliminieren. Die Proteine verantwortlich für diese Immunreaktion sind gemeinsam bekannt als die inflammasome, ein multiprotein intrazelluläre komplexe, die gebildet werden können, die von verschiedenen Proteinen und resultiert in der Bildung von verschiedenen proinflammatorischen Molekülen. Die Forscher fanden heraus, dass das vermittelnde in den chikungunya-virus-Infektion ist das Enzym caspase-1.
Der Befund wurde überprüft, in Experimenten mit Mäusen durchgeführt, in Zusammenarbeit mit Dario Zamboni, professor an FMRP-USP. Beide Nakaya und Zamboni sind mit dem angeschlossenen Zentrum für Forschung über Entzündliche Erkrankungen (CRID), eine von der Forschung, der Innovation und der Verbreitung Zentren (RIDCs) gefördert durch die São Paulo Research Foundation—FAPESP. Die CRID wird gehostet von FMRP-USP.
Sie fanden heraus, dass chikungunya-virus-Infektion, die in der genetisch veränderte caspase-1 knockout-Mäusen führte nicht zur Freisetzung von proinflammatorischen Molekül namens interleukin-1 beta (IL-1β), in der Erwägung, dass es in wild-Typ (nicht genetisch veränderten) Mäusen.
Nach der Identifizierung der gen-Signaturen, die das chikungunya-virus-Infektion, bei denen Tausende von Genen, deren expression verändert durch die Krankheit, die Gruppe verglichen die Ergebnisse mit denjenigen, die mit Proben von infizierten Patienten mit dengue-virus.
„Wir haben bemerkt, dass die gen-Signaturen bei beiden Erkrankungen ähnlich waren zu einem erheblichen Teil aber, dass einige Gene wurden spezifische chikungunya. Diese können weiter untersucht werden, die in Droge-Entwicklung-Forschung,“ sagte Nakaya.
In einer anderen Analyse, die Forscher verglichen die Genexpressionsprofile von infizierten Patienten mit dem chikungunya-virus mit dem Profil der Patienten mit rheumatoider arthritis, einer Autoimmunerkrankung, charakterisiert durch chronische Gelenk-Entzündungen.
„In diesem Fall war es unser Ziel, entdecken Sie den Unterschied zwischen arthritis verursacht durch das virus und Autoimmun-arthritis. Wir wollten zu identifizieren Gene, die bestimmte waren zu der viralen Infektion,“ Nakaya erklärt.
Kombinierte Analyse von drei gen-Signaturen zeigten, dass die 949 Gene waren bei rheumatoider arthritis allein, 632 in dengue allein, und 302 in chikungunya-allein. Sieben Gene, die verbunden waren mit allen drei Bedingungen: OAS1, C1QB, ANKRD22, IRF7, CXCL10, IFI6, und IFIT3.
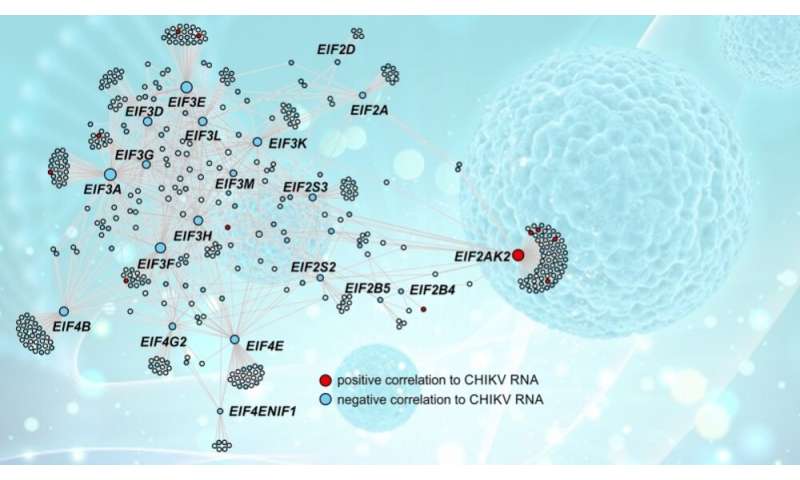
Die Forscher führten dann gen koexpression Analyse mit CEMiTool, ein software-Paket entwickelt von Nakaya mit FAPESP Unterstützung. Das Ziel dieser Analyse war es, zu verstehen, wie die Gene miteinander interagieren in einem komplexen Netzwerk, das existiert in jeder Zelle, bilden signalkaskaden und Stoffwechselwege.
„Wir identifizieren konnten acht Haupt-koexpression Module [Gene mit ähnlichen response profiles]. Wir ermittelten außerdem die Netzwerk-hubs—die Gene mit den meisten verbindungen und damit die vielversprechendsten Ziele zu erkunden, für die Arzneimittel-Entwicklung Zwecke,“ sagte Nakaya.
Die Daten in der Studie verwendet wird, werden sowohl die raw-Daten und Analyse-Ergebnisse werden in einem öffentlichen repository und kann heruntergeladen werden von jedermann kostenlos, wie er betonte, ebenso wie der code für das software-Paket bereits erwähnt, so dass andere die Ergebnisse reproduzieren.
„Unsere Forschung ermöglichte uns eine Liste der möglichen therapeutischen Ziele, und wir sind jetzt cross-referencing diese Ergebnisse mit einer Datenbank von aktiven verbindungen. Diese cross-referencing ist als getan, rechnerisch aber auf der Grundlage der experimentellen Daten zusammengestellt aus veröffentlichten Studien haben darauf hingewiesen, dass Drogen in der Lage sich mit dieser Gene von Interesse“, sagte Nakaya.