Neue Hinweise über die Herkunft der familiäre Formen der amyotrophen Lateralsklerose

Ein team unter der Leitung von brasilianischen Forscher Elis Eleutherio, professor an der Federal University of Rio de Janeiro in Partnerschaft mit Tiago Outeiro, Universität Göttingen, Deutschland, bedeutende Fortschritte im Verständnis der Konformation und Akkumulation von bestimmten Proteinen, die bei der amyotrophen Lateralsklerose (ALS).
„Wir glauben, dass protein-Ansammlung ist ein wichtiges Kennzeichen der ALS ist, und wir immer noch nicht verstehen, warum das protein benimmt und Aggregate während der Erkrankung“, erklärt Prof. Eleutherio.
Die ALS ist eine progressive und verheerende neurodegenerative Erkrankung, die sich auf 1 bis 3 Personen in 100.000, und es ist häufiger bei Menschen zwischen 55-75 Jahre alt. Die Krankheit betrifft in Erster Linie eine Bevölkerung von Neuronen, bekannt als ‚motor Neurone.‘ Die Patienten leiden unter irreversiblen motorischen Lähmung, und werden unfähig zu sprechen, zu schlucken oder zu atmen, wie die Krankheit fortschreitet.
Die meisten ALS-Fälle sind sporadisch, mit keine definierten genetischen Ursprung, und nur die Minderheit ist familiär, mit den bekannten damit verbundenen genetischen Veränderungen. Bestimmte familiäre Formen der ALS (fALS) sind im Zusammenhang mit genetischen Veränderungen in der gen-Kodierung für ein protein, bekannt als Superoxid-dismutase 1 (Sod1), dazu führen, dass Veränderungen in der Faltung und Funktion des proteins.
Die Studie, veröffentlicht in der Zeitschrift Proceedings of the National Academy of Sciences (PNAS), erlaubt Wissenschaftlern zu verstehen, die Interaktion zwischen dem normalen und dem mutierten protein, das bewirkt, dass die Veränderung von protein-Akkumulation in der Zelle, sondern beeinträchtigt auch die Funktion der Sod1-proteins, damit einen Beitrag zur Entwicklung der Krankheit. Für die Gruppe, diese Entdeckung eröffnet neue Perspektiven für die Behandlung von ALS.
Sod1 ist ein protein, das eine Rolle spielt, ist unter anderem der Schutz gegen oxidative Schäden in unseren Zellen. In einigen Fällen ALS, veränderten Sod1-protein sammelt sich im inneren, neuronalen Zellen und die Forscher glauben, bewirkt, dass Schäden an den Neuronen, führt zu deren Tod. Wichtig ist, normale Sod1 Proteine, die in sporadischen Fällen ALS, kann auch misfold und akkumulieren, was darauf hindeutet dies ist ein zentrales problem in der ALS.
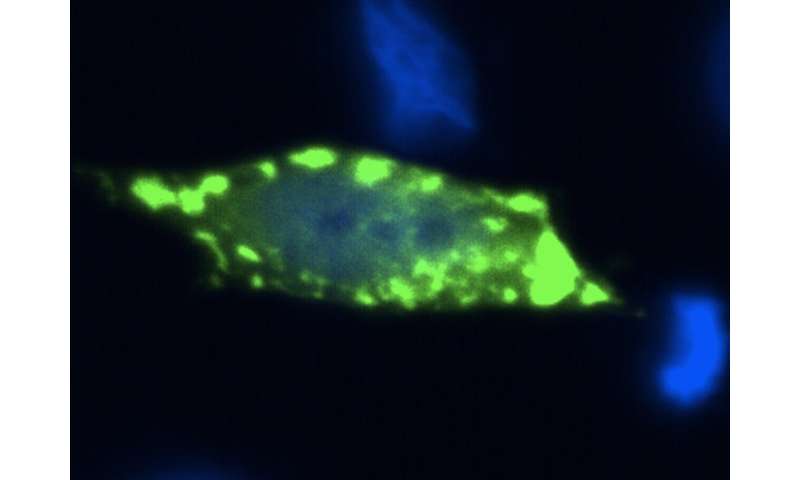
In der Studie, verwendeten die Forscher einfache experimentelle Modelle, wie Bäckerei Hefe verwendet, um Bier, Wein und Brot, und die menschlichen Zellen, um besser zu verstehen, den Kontext der protein-aggregation in der Krankheit. Sie verwendeten auch eine Strategie, die imitiert den genetischen Zusammenhang der fALS, wo die meisten Patienten tragen eine Kopie von der normalen Sod1-protein, und eine Kopie der Durchführung einer genetischen Veränderung. „Bei Patienten, denken wir, dass das Vorhandensein eines mutierten Kopie des Sod1 ändert sich das Verhalten der normalen kopieren“, erklärt Dr. Aline Brasil, der erste Autor der Studie.
„Durch die Nutzung von neuartigen genetischen manipulation tools und kraftvollen molekularen bildgebenden Verfahren, die es ermöglichen die direkte Visualisierung der protein-komplexe in der Zelle (eine Technik, bekannt als BiFC), waren wir in der Lage, zu erkennen, ‚hetero-komplexe‘ gebildet, die durch die normalen und anormalen (mutant) Sod1-protein“, sagte Prof. Outeiro, Leiter der deutschen Mannschaft, nahmen an der Studie Teil.
Die Forschung eröffnet neuartige Perspektiven für die therapeutische intervention, die die Autoren hoffen, auch weiterhin zu erkunden, in Naher Zukunft, wie die spezifische Entfernung des mutierten Sod1-proteins.