Biophysiker entdecken Sie, wie Sie ‚Australian‘ mutation führt zum Ausbruch der Alzheimer-Krankheit


Ein team von Wissenschaftlern des Moskauer Instituts für Physik und Technologie (MIPT) und Shemyakin-Owtschinnikow Institut für Bioorganische Chemie (IBCh, RAS) untersucht eine angeborene genetische mutation zu entdecken, die Allgemeinen molekularen Mechanismen, die möglicherweise führen sowohl zu frühen Beginn der Alzheimer-Krankheit und der form der Erkrankung, die durch altersbedingte Veränderungen im menschlichen Körper. Das Verständnis dieser Mechanismen ist notwendig für die Entwicklung neuer zielgerichteter Behandlungen für diese neurodegenerative Erkrankung, die immer mehr weit verbreitet entwickelte Länder “ Alterung der Bevölkerung. Die Ergebnisse der Studie wurden veröffentlicht in ACS Chemical Biology.
Demenz ist ein Syndrom, in dem es eine Verschlechterung in Gedächtnis, denken, Verhalten und die Fähigkeit, alltägliche Aktivitäten. Alzheimer-Krankheit ist die häufigste form der Demenz und kann dazu beitragen, die 60-70% der Fälle, nach Angaben der WHO fact sheet. Das macht die Demenz eine gesundheitspolitische Priorität, mit erheblichen Mittel zu kämpfen, die die Regierungen und Pharma-Unternehmen. Prominente Politiker wie Margaret Thatcher und Ronald Reagan wurden die betroffenen mit Alzheimer-Krankheit in Ihren späteren Jahren. Alzheimer-Krankheit ist am häufigsten bei Menschen über dem Alter von 65 sind, aber Menschen im Alter von 40 oder noch jünger sind, manchmal diagnostiziert als gut. Etwa 10-15% der früh auftretenden Fälle sind verursacht durch die erblichen Veranlagung. Integrierte Studien von erblichen oder „familiäre“ Mutationen können Forscher geben einen Anhaltspunkt über die wichtigsten Mechanismen der Alzheimer-Krankheit Pathogenese, insbesondere seine ersten Schritte.
Alzheimer-Krankheit ist assoziiert mit der Akkumulation pathogener amyloid-β-Peptide in der amyloid-plaques in Hirngewebe. Diese Peptide sind kurze (etwa 40 Aminosäuren) Fragmenten des amyloid-precursor-protein (APP), der sich durch die Membran von Hirnzellen. APP-protein wird gespalten durch verschiedene Enzyme, die als Teil der Neuronen-Aktivität. Die enzymatische Spaltung der „großen“ APP-proteins (biologische Funktion ist noch nicht vollständig verstanden) durch β – und γ-sekretase-Enzymen produziert amyloid-β-Peptide, die in kleinen Mengen sind wohl nötig für die Aufrechterhaltung der Gehirnfunktionen. Jedoch γ-sekretase schneidet das APP-Kette (innerhalb der Neuronen-Membranen) in aufeinander folgenden Fragmenten von leicht unterschiedlicher Länge, so dass eine relativ „pathogene“ und „apathogene“ Formen des amyloid-β-Peptiden. Die wichtigsten pathogene form besteht aus 42 Aminosäureresten (Aß42), während die weniger pathogene form besteht aus 40 Rückstände (Aß40). Die Aß42/Aß40-Verhältnis bei gesunden Menschen ist nicht hoch, stehen ungefähr eins bis neun. Eine höhere Aß42/Aß40-Verhältnis deutet auf eine übermäßige Produktion von Aß42 führt zu der neurodegenerativen Erkrankung. Die Forscher testen derzeit die Hypothese, dass amyloid-β-Peptide sind aktive Teilnehmer der angeborenen Immunität des menschlichen Nervensystems und Ihre Produktion gesteigert werden kann, verursacht durch verschiedene Entzündungen und Hirn-Verletzungen. Zur gleichen Zeit, eine Menge von familiären Mutationen im Zusammenhang mit frühen Beginn der Alzheimer-Krankheit gefunden wurden, die in der Transmembran – (TM) – Domäne von APP.
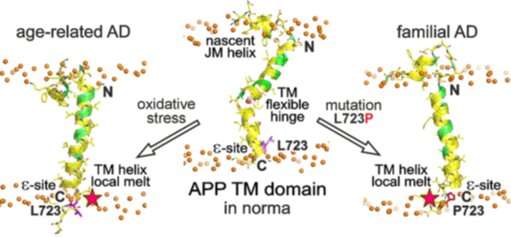
Diese Forschung hat zu der Studie der „Australian“ familiäre mutation (L723P) innerhalb der APP TM-Domäne, die die Ursache für frühen Beginn der Alzheimer-Krankheit. Die Wissenschaftler untersuchten die strukturell-dynamische Verhalten der Mutanten-APP-TM-Domäne gegen die wild-Typ durch die Hilfe von protein engineering, high-resolution nuclear magnetic resonance (NMR), und computer-Simulationen. NMR-Spektroskopie verwendet wurde, zu vergleichen mit dem Wildtyp-APP-Peptid mit seinem mutierten, indem solche Parameter wie „helicity“ der Aminosäuren-Polypeptid-Kette, seine Biegung und Flexibilität, sowie die Zugänglichkeit der Lipide und Wasser-Moleküle. Die Forscher entdeckten L723P mutation zu verursachen lokale Schmelzen der Letzte Zug von APP-TM-helix Domäne und auch die Festigung und Stabilisierung der Domäne in der Mitte der lipid-Membran. Abgesehen davon, es wurde festgestellt, dass die mutation erhöht die Erreichbarkeit der domain zu Wasser-Moleküle, die Verschiebungen der „Rahmen“ seiner Spaltung durch die γ-sekretase, damit die Umschaltung zwischen alternativen („pathogene“ und „apathogene“) die Spaltung Kaskaden. Dies führt zu wachsenden Aß42/Aß40-Verhältnis und die Allgemeine Konzentration von amyloid-β in Hirngewebe.
Wissenschaftlicher Mitarbeiter im Labor für das Altern und altersbedingte Neurodegenerative Erkrankungen, MIPT, Labor für biomolekulare NMR-Spektroskopie, IBCh, RAS, Eduard, Bocharov, kommentierte:
„Es geht ohne zu sagen, dass diese Studie berührt auf nur ein paar der Ursachen für die multifaktorielle Erkrankung, der Alzheimer-Krankheit. Die molekularen Mechanismen der Pathogenese sind, geforscht wird in zahlreichen Labors auf der ganzen Welt. Insbesondere, Besondere Aufmerksamkeit geschenkt Studium der „key player“—dem amyloid-precursor-protein, sowie deren enzymatische Spaltung durch sekretasen innerhalb der Neuronen-Membranen. Wir beschrieben eine Kaskade von Ereignissen in und um die Zellmembran als APP ist der Schnitt durch die γ-sekretase-Enzym-Komplex. Wir haben somit nur eine einzige „Australian“ – mutation zu offenbaren molekularen Mechanismen, die hinter der Pathogenese, die möglicherweise führen sowohl zu frühen Beginn der Alzheimer-Krankheit und die altersbedingte form der Krankheit.“